Recently, the team of professors Li Weimin and Xie Dan of West China Hospital published the results of multi-group research based on non-small cell lung cancer on cancer research.Lung cancer is one of the leading causes of cancer-induced death in the world. In recent years, lung cancer genome sequencing studies have revealed the relationship between some gene mutations and lung cancer, but there are still some lung cancer patients do not have obvious driving gene mutations, but have changed at the epigenetic level. Among them, the chromatin open region is an important epigenetic feature. The existence of open chromatin often means that the corresponding DNA regulatory elements are activated, thus regulating the expression of downstream genes. In view of the complex gene regulation model, simply observing the data changes of one dimension can not well analyze the occurrence and development of tumor. Therefore, it is of great significance to integrate multi-group data (genome, transfer group, epigenetic group) to reveal the complex gene regulatory network of lung cancer from many levels.
The research team integrates ATAC-seq technology, genome-wide sequencing technology and transcriptome sequencing technology to study the relationship of gene regulation in lung cancer. Through the analysis of chromatin open regions in 50 cases of non-small cell lung cancer, the researchers found that different pathological types of lung cancer have their own open region characteristics.
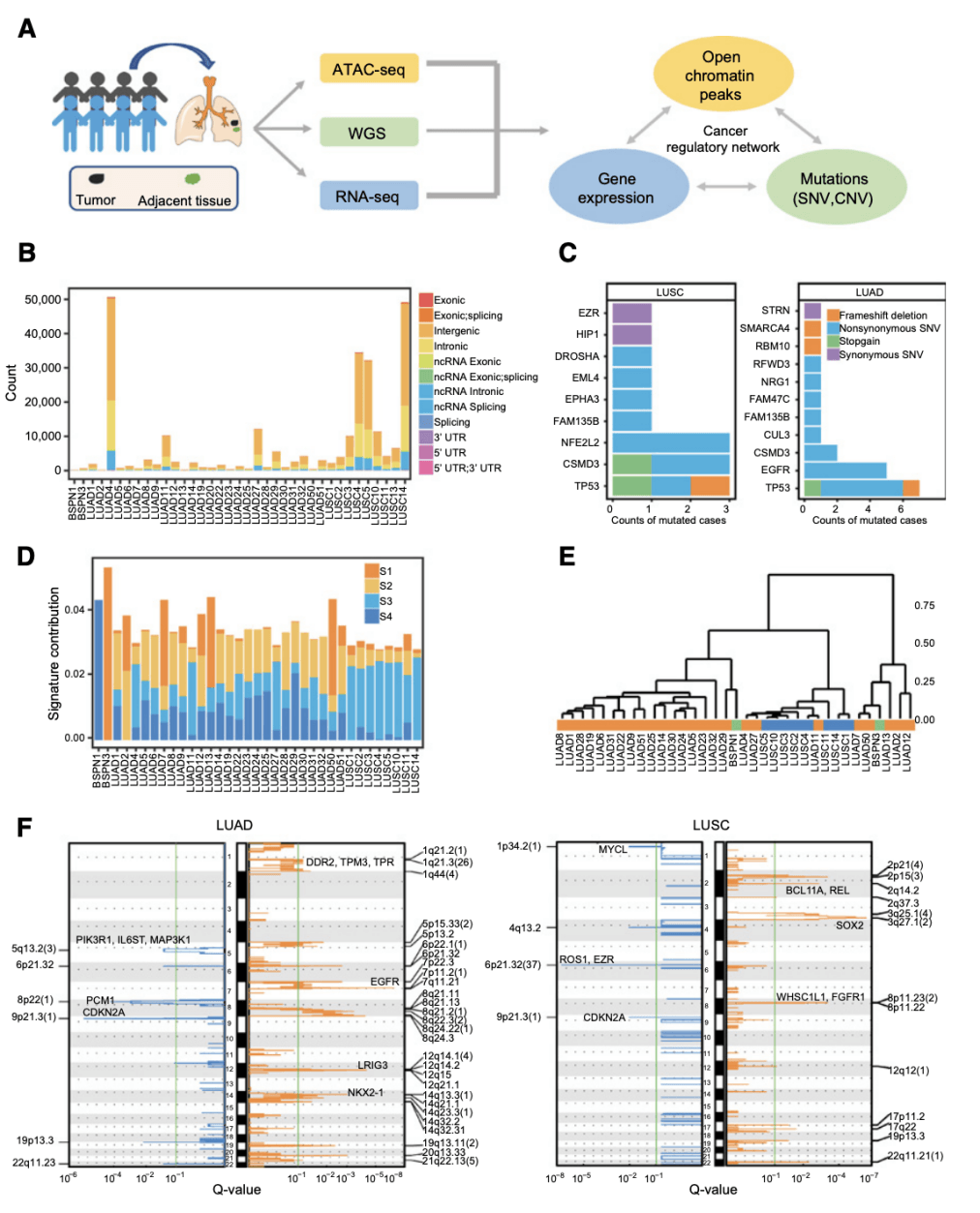
Figure 1. Overview of study and genetic landscape of patients with NSCLC.(Wang Z, et al. 2018)
From figure 1, we can see the researchers' overview of the design of the study. Barplot shows the number of somatic SNV located on different genomic features in each sample and the sample count of known cancer-related genes containing different somatic SNV and INDELs. The tree shows the results of hierarchical clustering of the samples based on four ab initio mutation markers, and the line chart shows significant copy number increase and loss.
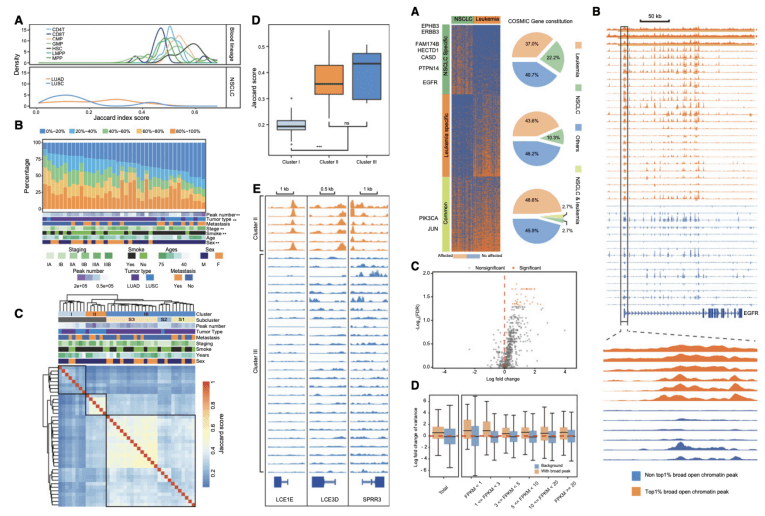 Figure 2. Integration of chromatin landscapes in NSCLC and the broad open chromatin region in NSCLC.(Wang Z, et al. 2018)
Figure 2. Integration of chromatin landscapes in NSCLC and the broad open chromatin region in NSCLC.(Wang Z, et al. 2018)
In addition, the researchers found a wide open distribution of (broad open chromatin peaks) in lung cancer samples. These special open regions range in length from 1kb to 180kb and are mainly enriched near the transcriptional initiation site of the gene. They not only show the unique distribution characteristics of cancer species, but also related to the degree of confusion of gene expression. The lung cancer driving genes near these regions, such as EGFR, ERBB3, etc., all showed large fluctuations in the expression of lung cancer in the samples. This phenomenon suggests that the wide open region may be involved in the process of tumor gene regulation disorder. Combined with chromosome structure variation data from DNA sequencing, the team also found that wide open regions were enriched in gene fragments with increased copies of the genome. At the same time, genomic copy number variation (CNV) also has a strong correlation with the open region signal and gene expression, that is, CNV will cooperate with the chromosome open region to affect the regulation of gene expression. In this study, the team observed a large number of genes affected by this expression regulation pattern.
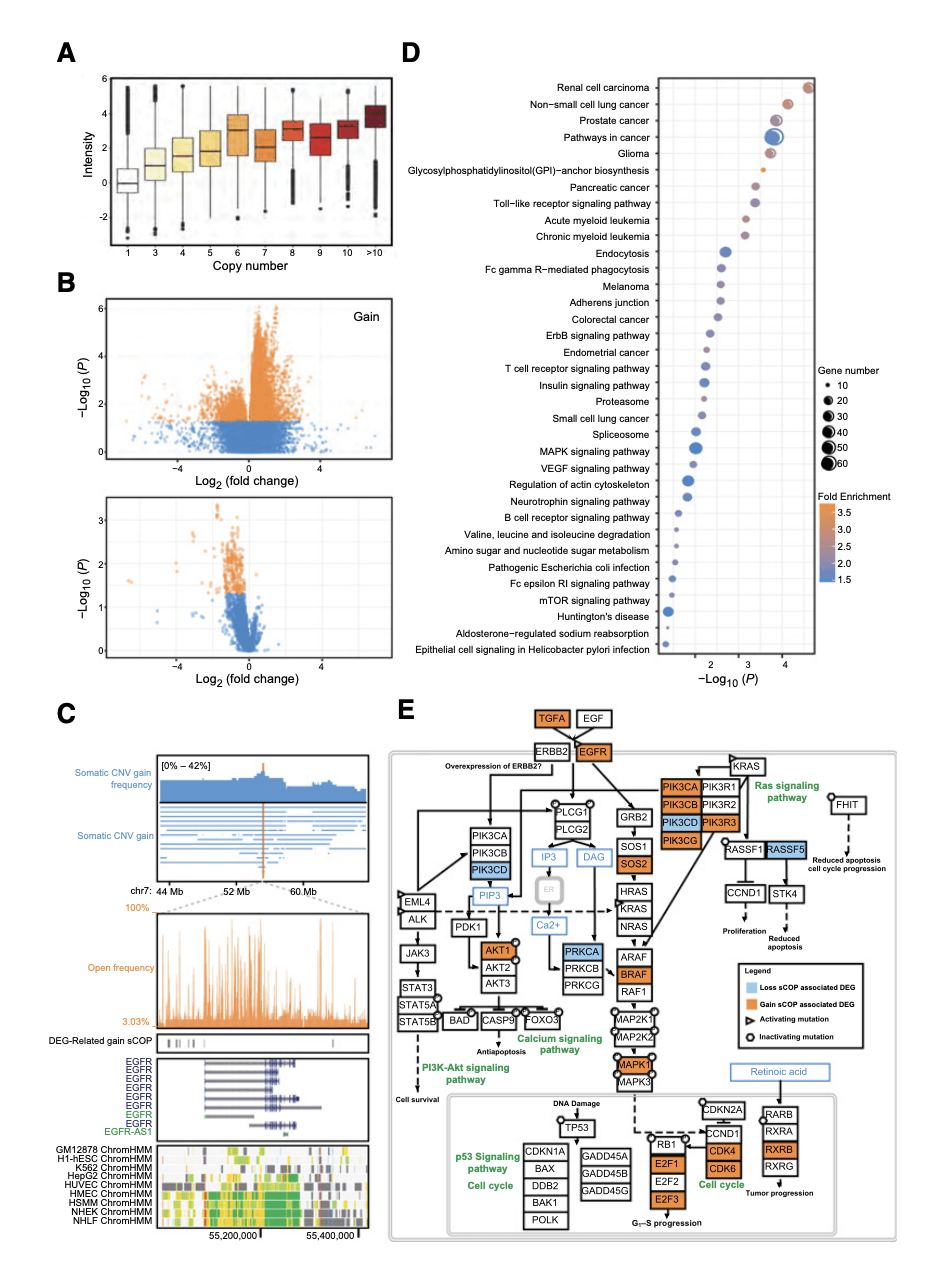
Figure 3. The association of somatic CNV, open chromatin peaks, and gene expression.(Wang Z, et al. 2018)
Multiple sets of data provide convenience for the analysis of gene regulation patterns of lung cancer. At the end of the paper, by analyzing the correlation between integrating different genomic information, the research team revealed the complex molecular regulatory network between genome sequence variation, large fragment DNA structural changes, open chromosome regions and gene expression changes. This regulatory model not only validates some of the results of previous genome-wide association analysis of lung cancer (GWAS), including some SNP sites related to the regulation of ROS and GSTT1 gene expression. The research team also found that a large number of genome segments located in non-coding regions play an important role in the regulatory network, and combined with bioinformatics methods to predict the gene regulation patterns and related transcriptional regulatory factors of these DNA fragments, which were originally regarded as "dark matter", and verified the function of some sites through lung cancer cell line experiments.
In this study, multiple sets of data were integrated to reveal the genomic regulatory network of non-small cell lung cancer, and cutting-edge genomic methods could be used to identify different subtypes of lung cancer. twenty-one quantitative trait loci (joint-QTLs) related to the open region of the genome and the regulation of transcriptional expression were found, and further confirmed the model that the loci related to lung cancer risk were involved in the genomic regulatory network proposed in the GWAS study. These regulatory sites located in non-coding regions can be used as targets for lung cancer diagnosis and drug design to guide the accurate treatment of lung cancer.
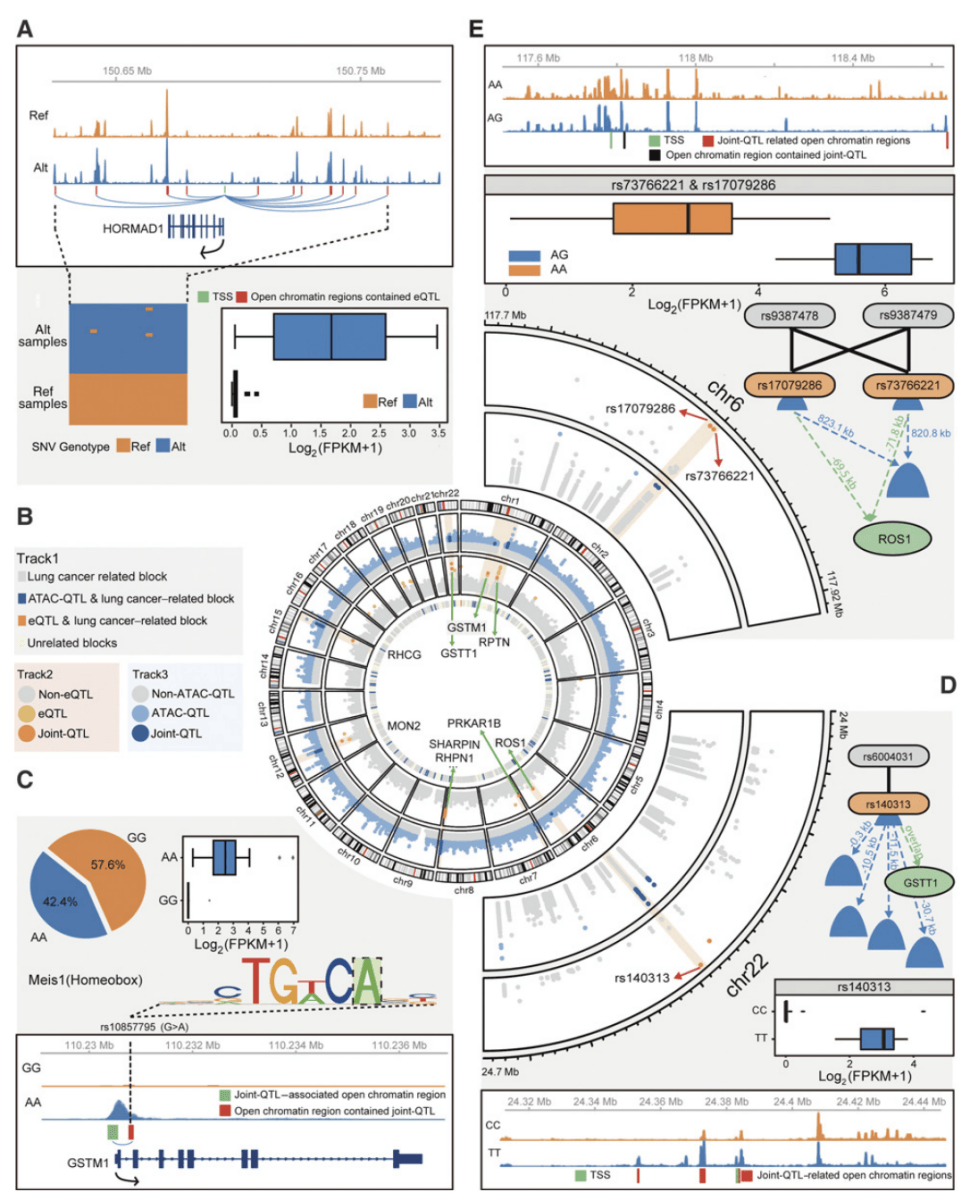 Figure 4. Molecular regulatory network of lung cancer based on multi-group data(Wang Z, et al. 2018)
Figure 4. Molecular regulatory network of lung cancer based on multi-group data(Wang Z, et al. 2018)
In this study, multiple sets of data were integrated to reveal the genomic regulatory network of non-small cell lung cancer, and cutting-edge genomic methods could be used to identify different subtypes of lung cancer. twenty-one quantitative trait loci (joint-QTLs) related to the open region of the genome and the regulation of transcriptional expression were found, and further confirmed the model that the loci related to lung cancer risk were involved in the genomic regulatory network proposed in the GWAS study. These regulatory sites located in non-coding regions can be used as targets for lung cancer diagnosis and drug design to guide the accurate treatment of lung cancer.
Creative Proteomics has an excellent team of experts to provide you with customized multi-group oncology-related experimental design and experimental solutions, so that you can experience the highest quality, the most advanced and the most satisfactory service.Help scientists in the field of cancer research to produce quick results and good results, so as to promote scientific and technological innovation. Creative proteomics, multi-layer assemblage customization service experts to help you with your scientific research!
Reference
1. Wang Z, Tu K, Xia L, et al. (2018). "The Open Chromatin Landscape of Non-small Cell Lung Carcinoma". Cancer research, 3663.
* For Research Use Only. Not for use in the treatment or diagnosis of disease.